報道
日前,FDA發布電子格式提交臨床研究用新藥安全性報告指南草案,
要求申辦者在指南最終版發布24個月后,向FDA不良事件報告系統提交IND的嚴重和意外可疑不良事件安全報告
。做出這一規定的原因何在?具體涉及到哪些提交?

改變現行提交方式,改進審查、跟蹤效率
按照現行要求,FDA要求這些報告都以PDF格式的電子通用技術文件提交。但對這些文件的審查和跟蹤效率低下,耗費大量人力。因此,
FDA要求將這些重要的安全信息作為結構化數據元提交給FAERS,有助于提高 FDA 審查和跟蹤臨床試驗期間發生的潛在安全信號的能力
,并為申辦方提供符合ICH E2B要求以及向其它監管機構報告的要求報告格式。對相關提交要求的轉變,是FDA具體落實《聯邦食品、藥品和化妝品法案》第745A節規定的電子提交要求工作的一部分。
FDA表示,申辦方目前還可以繼續通過eCTD提交相關的IND安全報告;但在要求生效之前,申辦方可以自愿向FAERS提交報告。但最終版指南一旦生效,FDA將要求IND安全報告的提交符合新指南的規定,取代2019年1月頒布的《按照eCTD規范提交部分人用藥品申請和相關提交》行業指南的相關規定。

依據指南規定,
申辦方必須通過 FDA 的電子提交網關或安全報告門戶 , 向FAERS 提交相關的 IND安全報告
。
通過FAERS提交IND安全報告所涉范圍
新公布的指南草案,不適用于嚴重和意外疑似不良事件之外的IND安全報告
,諸如詳細說明來自于其它研究的結果的報告,來自于實驗動物或體外檢測的結果,或嚴重疑似不良反應發生率增加的報告。此外,FDA表示,盡管FDA鼓勵申辦方以電子方式提交,非商業性IND的提交,將豁免第745A節規定的電子提交要求。
除了指南草案之外,作為指南草案的補充,FDA 還發布了單獨的技術符合性指南,詳細說明應以eCTD格式提交給FDA的個案安全報告 ,以及提交給 FAERS 的 IND 安全報告應采用的格式。
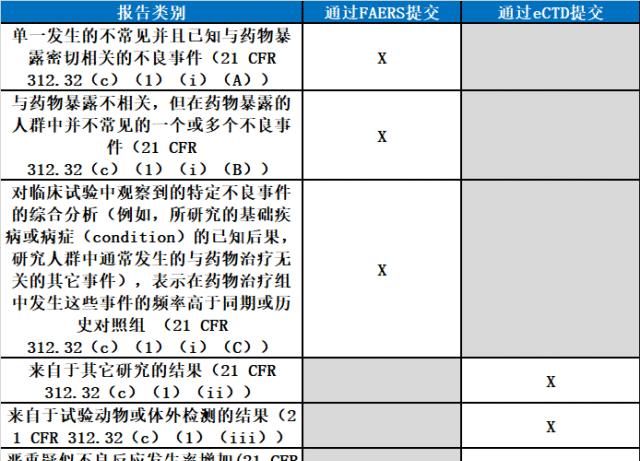
IND安全報告的類別與提交方式
FDA正在就這份指南草案征詢醫藥行業和利益攸關方的建議與意見,征詢日期截止于今年年底。
FDA新藥審評程序包括新藥臨床試驗申請IND申報和新藥申請NDA申報兩個過程,申請人在完成新藥臨床前研究后,便可向FDA提出IND申請,若FDA在收到后30天內未提出反對意見,申請人便可自行開展新藥臨......
2019年12月9日,藥明巨諾中國(下稱“藥明巨諾”)今日在第61屆美國血液病學會(ASH)年會上公布其首個在研產品JWCAR029治療成人復發/難治B細胞非霍奇金淋巴瘤(R/RB-NHL)的I期臨床......
昨日,Curis宣布,美國FDA已經接受了該公司產品CA-170的新藥研究申請(IND)。總部位于美國馬薩諸塞州萊克星頓的Curis是一家專注于人類癌癥藥物研究和開發的生物技術公司。CA-170是一劑......
創新藥研發是一個探索性的研究過程,是由未知開始,基于未被滿足的臨床需求,開展藥物篩選與發現的研究工作。因研發基礎不同,創新藥與仿制藥具有完全不同的研發路徑,因此其CMC申報也不相同。CMC指的是化學、......
時光飛逝,以前所學的專業知識似乎都還給老師了;還不到三年,初入職場時的興奮與激情似乎在悄悄地淡去。。。 &......





