五、EP05精密度實驗
EP05中提出的“批內不精密度”、“批間不精密度”的實驗方案,是較客觀地反映真實不精密度的估計。可惜,國內大多實驗室并沒有這樣去做。期望大家花點精力,學習EP05實驗方案。
目前EP5文件已經出了第三版(EP5-A3),內容較以往的版本要更加完整。但是,也使臨床實驗室在全面實施中引入了更多的困難。我正在學習中。因此這里,我還是按照EP5-A文件的內容做一個概要介紹。
1、EP05的精密度實驗方案主要做法。可以概括成幾句話:每天做2 批、每批做2份、連做20天。每批的兩個結果間差異,就是對該樣品早該批檢測不精密度的具體表現。每天上、下午各做一批,20天就有40批。也即最后的數據為20天、40對、80個。將40對結果間的不精密度差異合在一起,將得到較客觀的“批內不精密度”的估計。
每批做雙份,每天做兩批。將每批結果求均值,它代表這批檢測的平均水平。一天內兩批結果間的差異,代表了每天檢測“批間”的不精密度差異。將20天的“批間”差異合在一起統計,得到了20天內批間不精密度的估計。由于這一的估計必然包含了批內不精密度的影響,通過方差估計的計算,可以得到批間不精密度的估計。
每批做雙份、每天做兩批,則兩批4個結果的均值(也可以是各批結果均值的均值),代表當天對該樣品檢測的平均水平。
一共檢測了20天,有20個每天的檢測均值。將這些均值求一個總均值(也可以將80個結果去求均值)。所有每天均值對于總均值離散的程度,就是實驗室“天間”的不精密度估計。將批內不精密度的標準差、批間不精密度標準差、和天間不精密度標準差以方差形式疊加,得到這20天對該實驗樣品、在實驗的檢測系統下、在該檢驗操作人員實施下,得到的精密度估計。多年前將這樣的估計稱為“總的不精密度”估計,以后認為這樣不妥當,改稱為“實驗室內”不精密度(withinlabolatory imprecision)。現在對于實驗室內不精密度的估計,已經包括了在實驗室范圍內,檢測相同分析物的不同檢測系統的精密度實驗對不精密度估計綜合,成為該實驗室該分析物的實驗室內不精密度。
這樣認真的EP05實驗方案,是否很完美地反映實驗室在該分析物下的真實不精密度水平了嗎?30年前,這些起草EP05文件的專家就已經想的很多、很遠。他們是臨床實驗室的專家、質量管理專家、統計學專家、臨床專家、和行政管理官員等的完整組合。
2、起草專家首先考慮的是該文件的目的。
本文件是為體外診斷(IVD)設施廠商和臨床實驗室方法的開發者,他們期望建立他們方法的精密度能力。也是為這些方法的用戶,他們期望確認性能聲明的驗證,或簡單地要求度量他們自己的精密度。自動檢測程序的用戶期望僅使用最簡單的方案去確認廠商對精密度聲明的驗證的,應按照最近編輯的EP15-A2–用戶展示精密度和準確度性能。該導則對這些情況是完全通用的,因為他們包括考慮精密度估計可靠性的目標。
3、在精密度性能確認和驗證上的要求有區別。
針對我國大多實驗室均為“已經修改了原廠商檢測系統組成”的情況,所以,看來不是簡單地去驗證性能,應該是去確認自己性能(即:去建立實驗室自行組合的檢測系統性能)、或應該是建立這樣新組合的檢測系統的分析性能。若這樣,鼓勵使用EP05做法去確認精密度性能。
4、EP05的實驗設計,著重看來還是考慮實驗時間長短對精密度性能的影響。已經考慮到許多因素會影響精密度的結果。所以,在設計方案時,在EP5-A前文件上已經規定了:在一臺儀器上、一個批號的試劑、一個批號的校準品、一個操作人員的條件下,得到的不精密度水平。在這樣的條件下,實驗時間成為最重要因素去區分不同精密度的結果。
實驗設計沒有合并特別單獨估計其他可能的變異重要來源,如校準品或試劑批號的差異、或技師/操作人員的差異;但是,廠商應包括這樣的因素,以及在不同場地的設施間的變異。影響精密度的其他因素,如樣品準備、檢測材料穩定性、交叉污染、和漂移等被包括在本方案中作為實驗室內部精密度的來源,但沒有分別估計。
在完整方案中會使用一個批號的試劑和校準品,但結果的解釋(在合適時明確標記)必須包括這個事實,因為這樣的結果會低估長期實驗室內(或設施內)精密度。引入多批這些物質將增加觀察的變異,盡管實驗沒有允許分別估計這些因素的影響,但會較好地反映檢測系統在實際使用中的真實精密度性能。
上述內容明確告訴我們,對某個檢測系統的精密度性能估計時,越與日常檢驗工作接軌的,估計的精密度越真實。不僅需要如EP05要求的,20天、40批、80個結果的做法;而且還應像平時那樣:考慮多個操作人員輪班操作的、換用不同批號試劑和/或校準品批號校準、重新校準等,將這些日常的變異因素加到對精密度估計的影響,得到真實精密度水平。但決不可將任何更換試劑廠商、隨意調整計算因子等不規范行為加進去!
六、驗證實驗室精密度性能
當前的實驗室管理,要求實驗室在使用檢測系統檢測患者樣品、發出檢測報告前,必須驗證檢測系統的分析性能,確認實驗室分析性能符合廠商說明書中的性能聲明(Claim)。問題是:廠商在建立分析性能中必須要比臨床實驗室的常規檢測嚴密。為此,在EP05文件中對廠商建立性能提出了建議。可惜,全世界乃至我國,有那么多的試劑、儀器、和組合檢測系統,在國內銷售前均被我國的CFDA批準準許銷售。可是,每家廠商在建立分析性能的嚴密性上太有問題了。
按照美國政府的認識,無論FDA對體外診斷產品的管理,還是CLIA對臨床實驗室的管理,均代表政府行為。因此,實驗室使用的體外診斷產品必須得到FDA批準;實驗室在使用前必須按照CLIA要求對分析性能驗證(驗收)。驗證指標就是廠商的性能聲明!因此廠商的性能指標成為國家要求實驗室“遵守”的!在驗證中,我們實驗得到的精密度性能如何與廠商聲明的精密度性能比較呢?
現在的做法很簡單:在與廠商精密度指標采用的分析物濃度幾近一致的條件下,只要實驗室得到的標準差小于廠商的指標,該分析物項目的精密度性能被驗證認可!我們的實驗室管理部門從來沒有考慮:廠商說明書中的精密度指標是否符合要求?實驗室的實驗方案是否符合要求?廠商大多也是選擇方便的做!而且為了說明它們產品的質量優秀,數據會選擇偏小的報告。
因此,實驗室越是認真實驗,得到的精密度標準差數據越客觀,也許得到驗證的機遇也越小!所以,也沒有實驗室愿意按照EP05文件要求實驗,更不要說要如實進行。這樣的做法實在是當今的嚴重弊病!也因為如此,在與廠商精密度指標比較時,兩個不確切的精密度比較,很難說明究竟哪個精密度更真實!
為此,在EP05文件專家中,很早就認識到:單單將兩個標準差放在一起在數量上去比較是不可靠的。注意,這些比較是一個濃度下的點的比較!影響可靠性大小的最后,在廠商沒有說明實際得到標準差的實驗方案、但又獲得政府批準可用的情況下,只能假設廠商標準差的自由度為無窮大!
而實驗室的驗證實驗也做的很簡單,導至得到的精密度性能只能是“真實”標準差的一個很粗糙的抽樣,也即不可靠的因素很多。因此與廠商精密度指標比較時,必須調整檢測的自由度(degreeof freedom)。使實驗室的標準差與廠商標準差比較大小的指標,應在相應估計的自由度下去判斷。
在檢查用戶EP05和EP15的精密度結果是否與廠商“聲明”的一致上,CLSI導則創建了卡方檢驗,而不是F檢驗,這是依據以下的考慮。在廠商說明書(Instructionfor Use,IFU)的精密度表中,“聲明”代表了有關行政機構批準的標記(Labeled)標準差。[我對這段話的理解是:廠商的標準差經FDA批準后,已經成為實驗室必須要實現的目標。]
但是,廠商沒有明確說明,它們的精密度具體是怎樣實驗得到的。也即不可能確定與廠商估計有關的自由度,正如應用F檢驗要求的。也沒有任何廠商在進行重復性實驗中,是否進行多個試劑批號、多個儀器的實驗。缺少這個和其他信息。
用戶簡直沒有任何選擇去處理廠商說明書的值;而用戶必然處于需要去評估估計自由度的信息。再者,正如前述,實驗室本身估計的標準差也有很多不足的地方。一個做法是調整比較時的自由度。一般來說,自由度越小(相當于重復檢測次數),得到的抽樣標準差值具有的可信性較差,即標準差的可信區間很大。
專家介紹,F檢驗的自由度完全由比較的兩個標準差實驗自身各自重復次數決定,沒有可以調整的做法,所以,在統計上為統計功效上能力較差。相比之下,卡方檢驗(χ2)在比較標準差差異上具有較好的統計功效。這也是我以往都不懂的地方,至今依然非常模糊。
請教了工業上的有關精英,他們告知在統計上確實有使用卡方檢驗比較標準差的內容。
在前述實驗得到的精密度估計,應與設施精密度的性能聲明進行比較。應使用以下敘述的卡方(χ2)統計。為使用這個方法,性能聲明被表示為一個點的估計(即,一個標準差)。重復性和綜合精密度的估計應分別進行比較。
性能聲明標準差(
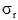
)須注明。卡方檢驗使用了用戶和廠商的重復性估計二者的平方。必須知道(即用戶估計的批內方差)與自由度個數的關系。在本方案敘述的實驗中,將具有用來計算批內標準差的數據成對(批內重復檢測)那樣大的自由度。這樣,這將等于實驗中的批數,以R注明。檢驗涉及的計算如下:
檢驗涉及的計算如下:
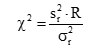
式中:
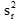
為用戶估計的重復性變異方差;
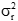
為廠商重復性變異聲明的方差;
R為批的總數(為 的自由度)。
因為重復性次數為80,所以R就是(80-1)。
應使用上述一樣的卡方檢驗,將實驗室內(設施內)精密度與廠商聲明進行比較。不像重復性估計,計算ST實際自由度涉及了復雜的計算。因為方案的結構,用戶不可假設所有觀察值是獨立的,去使用習慣估計的自由度(總觀察數減1)前的必要假設。以下公式對于ST的自由度T考慮了確實獨立性。
附:ST為實驗室內標準差。
卡方計算:
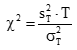
式中:
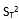
為用戶估計的實驗室內(設施內)標準差的平方值。
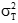
為廠商聲明的設施標準差的平方,或醫學上要求的標準差的平方。
T =
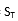
的自由度。
設:ME = (批內均方差,或重復性方差);
MR =
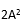
(所有批的均方差);
MD =
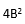
(所有天的均方差)。
I為實驗天數。
有關A和B,請同道閱讀EP文件。
T的計算較復雜:
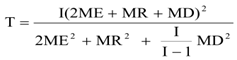
將計算的“自由度”T代入前述的公式,計算出實驗室內標準差與廠商聲明標準差比較的卡方值。在相應自由度下與臨界卡方值比較。計算卡方值小于臨界卡方值的,表示實驗室內標準差與廠商聲明標準差沒有顯著差異;反之,具有顯著差異。
七、總結
由于臨床實驗室檢驗的特殊做法,全球臨床實驗室對每個患者標本進行各個分析物的做法,在沒有特殊情況下,均對每個分析物只做單次檢測就發出報告。臨床和患者關注檢測結果是否滿意的認識,是再做一次的結果是否一樣。因此實現重復性是第一位的大事。千萬不可疏忽。
30余年前,美國臨床實驗室標準化委員會(NCCLS)已經考慮到多方需求和影響精密度的各個因素,寫就了EP05文件。完成一個重復性實驗方式多樣,為了患者樣品檢測結果的可靠性,必須充分考慮眾多影響因素,以適當方式得到可靠的精密度估計。
我在臨床實驗室領域內從事工作和學習50年了,但是還有許多方面需要我繼續努力學習的。精密度是我將其視為臨床實驗室的關鍵所在。幾乎所有的臨床檢驗問題大多均由此產生。因此始終關注和重視精密度性能是解決問題的基礎起點。我愿意與臨床實驗室的同道一起,為提高臨床實驗室地位去努力,依靠自身的提升,讓臨床和患者認可。臨床實驗室的地位只能靠自己去爭取。
在與CLSI交流中, Dr. Neill Carey專家告訴我: “在EP15-A2中的方法比較實驗,在EP15-A3中刪除。在EP-15A3中,我們不再比較兩個方法精密度的估計。”“我們做了實驗去評估在EP15(A2和A3)中一個方法的精密度。我們將觀察(計算)標準差與廠商聲明的標準差做比較。若觀察到的標準差超過廠商聲明的標準差,我們計算了廠商聲明標準差的確認限值,將觀察到的標準差與確認限值比較。如果觀察到的標準差超出確認限值,僅有5%的可能性,認為觀察到的標準差超出確認限值因機遇所致;即觀察到的標準差確實高于聲明的標準差。”這兩個文件已經正式發布。新的文件不再要求臨床實驗室對驗證中實驗精密度超出廠商聲明精密度,進行統計檢驗。這無疑是簡化了驗證程序。