DNA微陣列基因表達數據分析
本文利用Matlab及其生物信息學 工具箱提供的函數識別差異表達基因并利用基因本體論確定差異表達基因的生物學功能。
引言
包含寡核苷酸或cDNA 探針的微陣列可用來比較基因組尺度的基因表達譜,微陣列試驗的重要目的在于確定不同條件下,如兩種不同的腫瘤類型,是否存在統計顯著的基因表達量的變化進而確定差異表達基因的生物學功能。
本文利用一個公共數據集來說明計算過程,這個數據集包括42個胚胎中樞神經系統腫瘤組織(CNS, Pomeroy et al. 2002),樣本采用Affymetrix 公司出品的HuGeneFL基因芯片進行雜交 。
這些CNS數據集(CEL文件)可在CNS實驗網站獲得,42個腫瘤樣本包括10個10 個髓母細胞瘤, 10個橫紋肌樣腦膜瘤, 10個膠質瘤, 8個幕上原始神經外胚層腫瘤和4個正常人小腦,CNS原始數據集用魯棒多芯片平均(RMA)和GC魯棒多芯片平均(GCRMA)進行了預處理。
可以采用t檢驗和假發現率(FDR)來檢測不同腫瘤類型間差異表達的基因,還可以探索與顯著上跳基因相關的基因本體論術語。
載入基因表達數據
用Load命令加載MAT文件cnsexpressiondata包含三個DataMatrix對象,expr_cns_rma, expr_cns_gcrma_mle, and expr_cns_gcrma_eb,分別儲存用RMA和GCRMA(MLE和EB)預處理的基因表達值。
load cnsexpressiondata
在每個DataMatrix對象中,每行對應一個HuGeneFl芯片的探針集,每列對應于一個樣本,行名是探針集的ID而列名為樣本名,本文用expr_cns_gcrma_eb示例,當然也可以用其他對象。
調用get命令獲取DataMatrix對象的特征。
get(expr_cns_gcrma_eb)
Name: 'CNS gene expression data'
RowNames: {7129x1 cell}
ColNames: {1x42 cell}
NRows: 7129
NCols: 42
NDims: 2
ElementClass: 'single'
確定DataMatrix對象expr_cns_gcrma_eb中的基因和樣本的數目。
[nGenes, nSamples] = size(expr_cns_gcrma_eb)
nGenes =
7129
nSamples =
42
可以用基因符號來代替探針集的ID用于標記基因表達值,HuGeneFl芯片的基因符號在一個包含Java哈希表的MAT文件中。
load HuGeneFL_genesymbol_hashtable;
為hu6800genesymbol_hashtable變量創建一個基因表達值的基因符號的cell矩陣。
huGenes = cell(nGenes, 1);
for i =1:nGenes
huGenes{i} = hu6800genesymbol_hashtable.get(expr_cns_gcrma_eb.RowNames{i});
end
用DataMatrix的rownames方法將exprs_cns_gcrma_eb中的行名設成基因符號。
expr_cns_gcrma_eb = rownames(expr_cns_gcrma_eb, ':', huGenes);
基因表達數據的過濾
首先除去沒有基因符號的表達數據,如標成'---'的空符號。
expr_cns_gcrma_eb('---', :) = [];
在這個研究中很多基因沒有表達或在樣本間變化很小,這些基因需要用非特異性過濾除去。
用genelowvalfilter函數濾除絕對表達量值很低的基因。
[mask, expr_cns_gcrma_eb] = genelowvalfilter(expr_cns_gcrma_eb);
用genevarfilter函數濾除樣本間方差很小的基因。
[mask, expr_cns_gcrma_eb] = genevarfilter(expr_cns_gcrma_eb);
確定過濾以后的基因數目。
nGenes = expr_cns_gcrma_eb.NRows
nGenes =
5758
識別差異基因表達
現在可以比較一下CNS髓母細胞瘤(MD)和非神經源惡性膠質瘤(Mglio)之間基因表達值的差異了。
從42個樣本中提取10個MD和10個Mglio樣本數據。
MDs = strncmp(expr_cns_gcrma_eb.ColNames,'Brain_MD', 8);
Mglios = strncmp(expr_cns_gcrma_eb.ColNames,'Brain_MGlio', 11);
MDData = expr_cns_gcrma_eb(:, MDs);
get(MDData)
Name: ''
RowNames: {5758x1 cell}
ColNames: {1x10 cell}
NRows: 5758
NCols: 10
NDims: 2
ElementClass: 'single'
MglioData = expr_cns_gcrma_eb(:, Mglios);
get(MglioData)
Name: ''
RowNames: {5758x1 cell}
ColNames: {1x10 cell}
NRows: 5758
NCols: 10
NDims: 2
ElementClass: 'single'
通常t檢驗是檢測兩組變量之間顯著性差異的標準統計檢驗,對每個基因執行t檢驗以識別MD樣本和Mglio樣本基因表達值的顯著性差異,可以通過t得分的正態分位圖和t得分及p值的直方圖來研究檢驗結果。
[pvalues, tscores] = mattest(MDData, MglioData,...
'Showhist', true', 'Showplot', true);

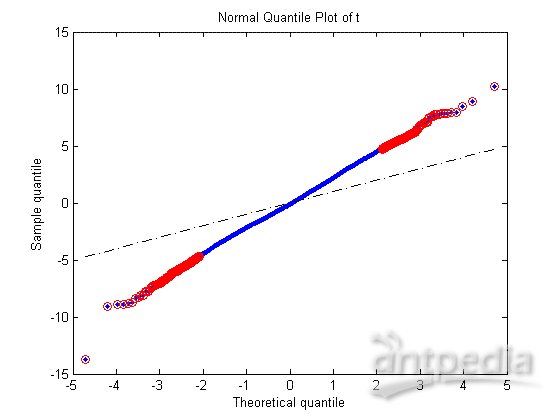
在所有的檢驗情形下都存在兩類誤差,當一個非差異表達的基因被標記為差異表達的基因時產生假陽性,而一個差異表達基因未能識別出來時產生假陰性,進行多重假設檢驗時,即用基因表達數據對上千個基因同時檢驗員假設時,每個檢驗都存在其特殊的假陽性率,或加發現率(FDR),FDR定義為假陽性基因數與總陽性數的期望之比(Storey et al., 2003)。
Storey-Tibshirani例程不僅計算FDR,還得出檢驗的q值,代表檢驗的最小FDR,因為FDR的估計依賴于多重檢驗的真實的空分布,這是未知的,通過交換基因表達數據矩陣的列的替換方法可用來估計真實的空分布(Storey et al., 2003, Dudoit et al., 2003),鑒于樣本數量,考慮所有的替換是不可行的通常在大樣本下只考慮一個隨機子集,本文利用統計工具箱中的nchoosek函數找出所有可能的替換數。
all_possible_perms = nchoosek(1:MDData.NCols+MglioData.NCols, MDData.NCols);
size(all_possible_perms, 1)
ans =
184756
調用mattest的PERMUTE選項進行替換的t檢驗,通過對基因表達數據矩陣MDData和MglioData的列進行10,000次替換來算出p值。(Dudoit et al., 2003)
pvaluesCorr = mattest(MDData, MglioData, 'Permute', 10000);
以0.05為p值的閾值確定具有統計顯著的差異表達的基因數目,由于替換檢驗結果不同可能會得到不同的基因數。
cutoff = 0.05;
sum(pvaluesCorr < cutoff)
ans =
2007
用mafdr對每個檢驗估計FDR和q值,pi0 是研究中真的空假設的總的分數,可以通過bootstrap或立方多項式擬合來估計模擬的空分布,也可以手動設置λ值來估計pi0 。
figure;
[pFDR, qvalues] = mafdr(pvaluesCorr, 'showplot', true);
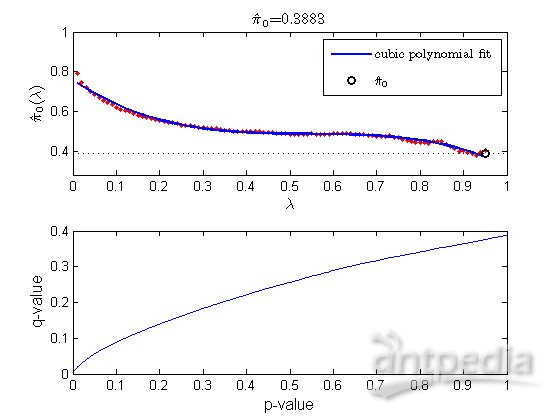
確定q值低于設定閾值的基因數,同樣,由于替換和bootstrap結果的不同可能得出不同的基因數。
sum(qvalues < cutoff)
ans =
1925
如果大量基因具有低的FDR表明兩組樣本具有生物學差異。
通過將mafdr函數的輸入參數BHFDR設為真可以調用Benjamini-Hochberg (BH)例程(Benjamini et al, 1995)估計FDR調節p值。
pvaluesBH = mafdr(pvaluesCorr, 'BHFDR', true);
sum(pvaluesBH < cutoff)
ans =
1275
可以將檢驗結果包括t得分, p值, pFDRs, q值和BH FDR校正p-values 存為DataMatrix對象。
testResults = [tscores pvaluesCorr pFDR qvalues pvaluesBH];
可以用DataMatrix對象的colnames方法將列名更新為BH FDR校正的p 值。
testResults = colnames(testResults, 5, {'FDR_BH'});
可以用sortrows方法對p-值pvaluesCorr進行排序。
testResults = sortrows(testResults, 2);
下面顯示檢驗結果的前23個基因,注意,由于替換測驗和bootstrap 結果的差異,最終結果可能和這里顯示的有所不同。
testResults(1:23, :)
ans =
t-scores p-values FDR q-values
PAFAH1B3 10.211 8.1924e-009 1.8315e-005 1.8315e-005
RAB31 -13.702 2.8016e-008 3.1316e-005 3.1316e-005
LRP1 -9.0742 1.6453e-007 0.00012261 5.3953e-005
KIAA0367 -8.9398 1.7404e-007 9.7272e-005 5.3953e-005
ID2B -8.8966 1.7782e-007 7.9509e-005 5.3953e-005
FBL 8.8668 1.8071e-007 6.7333e-005 5.3953e-005
PLEC1 -8.7961 1.8858e-007 6.0228e-005 5.3953e-005
PEA15 -8.7637 1.9306e-007 5.3953e-005 5.3953e-005
HNRPA1 8.4532 2.6546e-007 6.5942e-005 6.1939e-005
ID2B -8.4194 2.7705e-007 6.1939e-005 6.1939e-005
ITGAV -8.265 3.5525e-007 7.2201e-005 6.4346e-005
KIR2DL1 -8.1355 3.8039e-007 7.0867e-005 6.4346e-005
SPARCL1 -8.1246 3.8603e-007 6.6387e-005 6.4346e-005
PTOV1 7.9738 5.5184e-007 8.8123e-005 6.4346e-005
RBMX 7.9186 6.0384e-007 8.9997e-005 6.4346e-005
H3F3A 7.8816 6.2271e-007 8.7009e-005 6.4346e-005
DHX9 7.8439 6.6235e-007 8.7103e-005 6.4346e-005
NAP1L1 7.8241 6.6698e-007 8.2839e-005 6.4346e-005
C5orf13 7.8037 6.6868e-007 7.868e-005 6.4346e-005
ZAP70 7.7977 6.6897e-007 7.4778e-005 6.4346e-005
MS4A1 -7.7607 6.7364e-007 7.1715e-005 6.4346e-005
GPM6B -7.7605 6.737e-007 6.8461e-005 6.4346e-005
RNF113A -7.7548 6.7548e-007 6.5658e-005 6.4346e-005
FDR_BH
PAFAH1B3 4.7172e-005
RAB31 8.0657e-005
LRP1 0.00013896
KIAA0367 0.00013896
ID2B 0.00013896
FBL 0.00013896
PLEC1 0.00013896
PEA15 0.00013896
HNRPA1 0.00015953
ID2B 0.00015953
ITGAV 0.00016573
KIR2DL1 0.00016573
SPARCL1 0.00016573
PTOV1 0.00016573
RBMX 0.00016573
H3F3A 0.00016573
DHX9 0.00016573
NAP1L1 0.00016573
C5orf13 0.00016573
ZAP70 0.00016573
MS4A1 0.00016573
GPM6B 0.00016573
RNF113A 0.00016573
一個基因被認定是在兩組樣本間差異表達的應該具有統計學和生物學意義,本文比較MD和Mglio腫瘤樣本之間的基因表達值之比,因此上調基因表明在MD中高表達而下調基因在Mglio中高表達。
用p-值的–log10對生物學效應可以畫成火山圖,注意,通過火山圖用戶界面UI,可以交互式地改變p-值的閾值和變化倍數的限定并輸出差異表達基因。
diffStruct = mavolcanoplot(MDData, MglioData, pvaluesCorr)
diffStruct =
Name: 'Differentially Expressed'
PVCutoff: 0.0500
FCThreshold: 2
GeneLabels: {116x1 cell}
PValues: [116x1 bioma.data.DataMatrix]
FoldChanges: [116x1 bioma.data.DataMatrix]

按下Ctrl鍵的同時點擊基因列表中的基因名可以在圖中找到基因,如火山圖所見,小腦顆粒神經元細胞特異的基因ZIC 和NEUROD 上調,而星形和少突膠質細胞分化基因如SOX2, PEA15, 和ID2B,則下調。
確定差異表達基因數。
nDiffGenes = diffStruct.PValues.NRows
nDiffGenes =
116
獲得上調基因列表。
up_geneidx = find(diffStruct.FoldChanges > 0);
up_genes = rownames(diffStruct.FoldChanges, up_geneidx);
nUpGenes = length(up_geneidx)
nUpGenes =
75
確定下調基因數。
nDownGenes = sum(diffStruct.FoldChanges < 0)
nDownGenes =
41
用基因本體論(GO)注釋上調基因
可以通過基因本體論(GO)來注釋差異表達基因,從上面的分析結果中查找上調基因,從Gene Ontology Current Annotations下載人類注釋(gene_association.goa_human.gz),解壓縮并存入當前目錄。
找出上調基因號用于GO分析。
huGenes = rownames(expr_cns_gcrma_eb);
for i = 1:nUpGenes
up_geneidx(i) = find(strncmpi(huGenes, up_genes{i}, length(up_genes{i})), 1);
end
用geneont函數加載GO數據庫為MATLAB對象。
GO = geneont('live',true);
讀人類注釋文件,本文只看與分子功能相關的基因,故只需讀Aspect域設為“F”的信息,基因符號和對應ID是我們感興趣的域,在GO注釋文件中分別為DB_Object_Symbol和GOid域。
HGann = goannotread('gene_association.goa_human',...
'Aspect','F','Fields',{'DB_Object_Symbol','GOid'});
創建人類基因及相應GO術語的列表。
HGgenes = {HGann.DB_Object_Symbol}; % Homo sapiens gene list
HGgo = [HGann.GOid]; % associated GO terms
確定分子功能相關的人類注釋基因數。
numel(HGgenes)
ans =
74298
并非所有的HuGeneFL芯片上的5758個基因都有注釋,通過比較基因符號列表和GO中的基因符號列表可以看出其是否已被注釋,可以跟蹤注釋基因數和每個GO術語關聯的上調基因數。
m = GO.Terms(end).id; % gets the last term id
chipgenesCount = zeros(m,1); % a vector of GO term counts for the entire chip.
upgenesCount = zeros(m,1); % a vector of GO term counts for up-regulated genes.
for i = 1:length(huGenes)
idx = strncmpi(HGgenes, huGenes{i}, length(huGenes{i})); % lookup genes
goid = HGgo(idx);
goid = getrelatives(GO,goid);
% Update the tally
chipgenesCount(goid) = chipgenesCount(goid) + 1;
if (any(i == up_geneidx))
upgenesCount(goid) = upgenesCount(goid) +1;
end
end
可以用超幾何分布確定統計顯著的GO術語,對于每一個GO術語,p-值表示關聯的注釋基因由于偶然性獲得的概率。
gopvalues = hygepdf(upgenesCount,max(chipgenesCount),...
max(upgenesCount),chipgenesCount);
[dummy, idx] = sort(gopvalues);
report = sprintf('GO Term p-value counts definitionn');
for i = 1:10
term = idx(i);
report = sprintf('%s%st%-1.5ft%3d / %3dt%s...n',...
report, char(num2goid(term)), gopvalues(term),...
upgenesCount(term), chipgenesCount(term),...
GO(term).Term.definition(2:min(50,end)));
end
disp(report);
GO Term p-value counts definition
GO:0030528 0.00044 8 / 489 Plays a role in regulating transcription; may bin...
GO:0003700 0.00090 8 / 538 The function of binding to a specific DNA sequenc...
GO:0003677 0.00165 8 / 583 Interacting selectively with DNA (deoxyribonuclei...
GO:0003705 0.00422 6 / 351 Functions to initiate or regulate RNA polymerase ...
GO:0043169 0.00519 5 / 245 Interacting selectively with cations, charged ato...
GO:0015458 0.00559 1 / 1 ...
GO:0016249 0.00559 1 / 1 Functions to locate the position of ion channels ...
GO:0016974 0.00559 1 / 1 ...
GO:0005509 0.00968 7 / 564 Interacting selectively with calcium ions (Ca2+)....
GO:0030020 0.01069 2 / 30 A constituent of the extracellular matrix that en...
可以研究顯著的GO術語并選擇與具體的分子功能相關的術語來構建一顆包括其祖先的子本體論樹,用biograph函數來可視化顯示它,也可為節點著色,本文中紅色的節點是最顯著的,蘭色的節點最不顯著,由于人類基因注釋文件的頻繁更新可能返回不同的GO術語。
fcnAncestors = GO(getancestors(GO,idx(1:4)))
[cm acc rels] = getmatrix(fcnAncestors);
BG = biograph(cm,get(fcnAncestors.Terms,'name'))
for i=1:numel(acc)
pval = gopvalues(acc(i));
color = [(1-pval).^(1) pval.^(1/8) pval.^(1/8)];
set(BG.Nodes(i),'Color',color);
end
view(BG)
Gene Ontology object with 8 Terms.
Biograph object with 8 nodes and 9 edges.

從代謝通路中發現差異表達基因
通過KEGG's SOAP Web Service或將基因符號列表發送到KEGG's Web query tool可以從KEGG代謝途徑數據庫查詢差異表達基因的代謝途徑信息。
References
[1] Pomeroy, S.L., Tamayo, P., Gaasenbeek, M., Sturla, L.M., Angelo, M., McLaughlin, M.E., Kim, J.Y., Goumnerova, L.C., Black, P.M., Lau, C., Allen, J.C., Zagzag, D., Olson, J.M., Curran, T., Wetmore, C., Biegel, J.A., Poggio, T., Mukherjee, S., Rifkin, R., Califano, A., Stolovitzky, G., Louis, DN, Mesirov, J.P., Lander, E.S., and Golub, T.R. (2002). Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature, 415(6870), 436-442.
[2] Storey, J.D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc.Nat.Acad.Sci., 100(16), 9440-9445.
[3] Dudoit, S., Shaffer, J.P., and Boldrick, J.C. (2003). Multiple hypothesis testing in microarray experiment. Statistical Science, 18, 71-103.
[4] Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc., B 57, 289-300
6月10日,Mathworks稱,迫于美國政府的政策,禁止向哈爾濱工業大學、哈爾濱工程大學提供技術以及客戶支持服務。此后,這一言論被證實,兩校師生無法使用該公司旗下數據分析、圖像處理軟件Matlab。......
哈爾濱工業大學哈工大、哈工程的老師和學生們最近無法使用MATLAB了,這一消息迅速成為了人們關注的熱點。近日,在知乎等社交網絡上,有哈工大學生表示收到了正版軟件取消激活的通知,而在與MATLAB開發公......
10月21日,安捷倫科技公司宣布推出N9030APXA信號分析儀――AgilentX系列信號分析儀中性能最高的分析儀。PXA提供高達26.5GHz的頻率范圍,通過可選的測量功能和硬件可擴展性能夠確保當......
a=double(imread(‘圖片名稱.后綴’));size(a)xy=x*y;%根據矩陣大小求出圖片像素點個數,x,y分別是size(a)求得結果的前兩項數字r=a(:,:,1);g=a(:,:......