PCR實驗包括三大主要的熱循環步驟 ,在這個過程中需要多種必要的反應成分 ,通過對PCR的設置做相應的調整,便可以獲得一些特殊的實驗結果,如產率提高,特異性增強或反應時間縮短等。
常用于增強PCR擴增的特異性。該方法主要利用抗體、親合配體、適體或化學修飾物等酶修飾酶,來抑制室溫下的DNA聚合酶的活性。這種修飾使得在PCR體系配制階段引物與模板、引物與引物之前的結合能力降低,從而避免了非特異性擴增。由于DNA聚合酶在室溫下的活性被抑制,所以,熱啟動技術為在室溫下配制多個PCR反應體系提供了極大的便利,且不會影響特異性和擴增能力。當反應體系配制好后,在反應初始加熱階段或“熱啟動”階段,酶修飾物在高溫下(通常高于90 ℃)被釋放,使得DNA聚合酶被激活。具體激活時間和溫度取決于DNA聚合酶以及熱啟動修飾物的性質。對某些DNA聚合酶而言,有時激活和起始變性步驟可以合并為一步。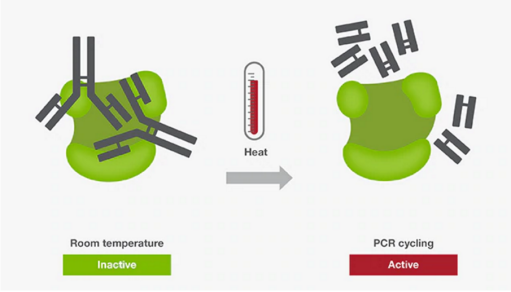
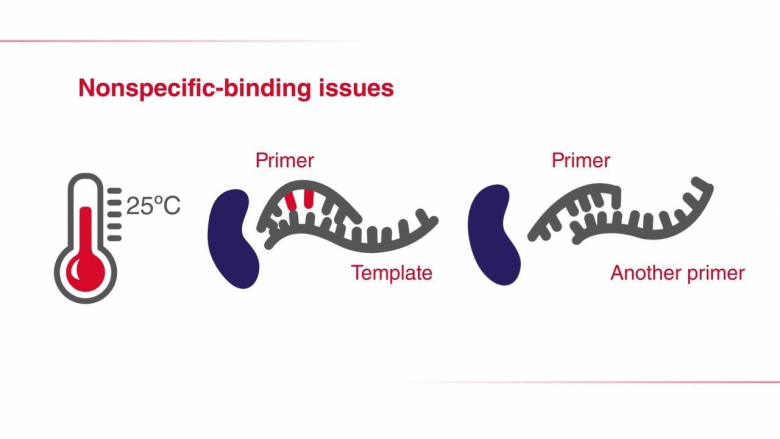
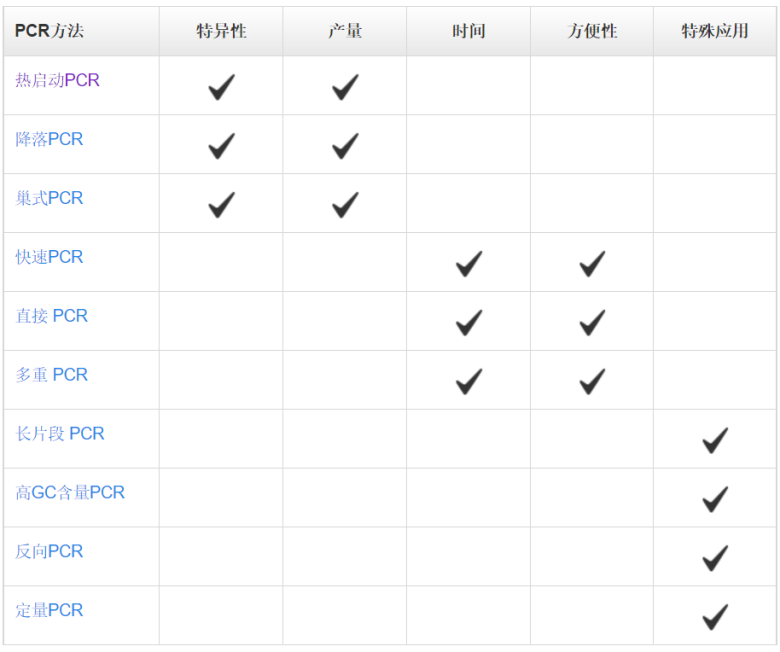
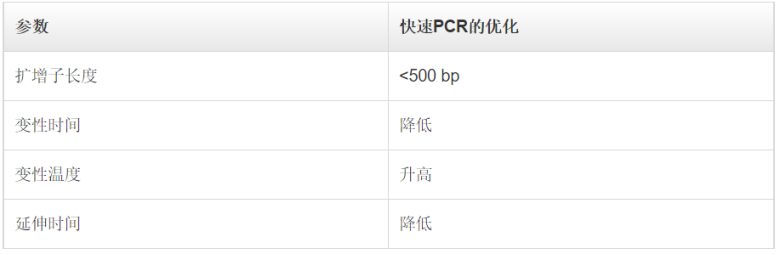
另一種提高PCR反應特異性的方法是調整PCR循環的參數。在降落PCR中,前面幾個循環的退火溫度設定為比引物的最高熔解溫度(Tm)再高幾度。較高的溫度有助于避免產生引物二聚體及非特異性引物-模板復合物的形成,因此可減少不希望出現的擴增。因此,在PCR起始階段提高退火溫度,可減少非特異性PCR產物,增加特異性擴增。需要注意的是,雖然較高的退火溫度能夠防止引物二聚體形成和非特異性引物結合,但同時也可能加劇引物與目的序列的解離,從而降低PCR得率。為克服這一問題,在最初幾個循環中,通常會將每個循環的退火溫度降低1°C,以獲得足量的目標擴增子。一旦退火溫度達到或“降落”至最佳溫度(通常比最低引物Tm低3-5°C),剩下的循環都維持此退火溫度。通過這種方法,在PCR過程中,所期望的PCR產物得到有選擇性的增加,同時保證很少或不發生非特異性擴增。巢式PCR是標準PCR的一種演變,其增強了反應特異性和目標擴增子的產量。在此方法中,需要設計兩對PCR引物:一對(外引物)在目標擴增區域的側翼,另一對(巢式引物)對應于待擴增的DNA區域。其中,外引物用于第一輪PCR,以擴增含有延伸側翼區域的區域。隨后,巢式引物用于第二輪PCR,并以第一輪PCR產物為模板。如果第一對引物(外引物)的錯配導至非特異性產物被擴增,相同的非特異性區域被第二對引物識別并繼續擴增的可能性非常小,所以通過第二對引物的擴增,PCR的特異性得到了提升。進行兩輪PCR的一個優勢在于:有助于從有限的起始DNA中擴增得到足量的產物。在快速PCR中,通過縮減PCR步驟所需時間來完成更快的擴增,且不會影響擴增產量和效率。快速循環條件尤其適用于具有高擴增能力的DNA聚合酶,這類聚合酶在每個結合中可引入更多的核苷酸。高合成能力 Taq 聚合酶所需的延伸時間僅占低合成能力Taq聚合酶所需時間的1/2至1/3,卻能維持較高的擴增效率。此外,如果引物的退火和延伸溫度相差無幾,則可將它們合并為一步,以進一步縮短PCR時間。這一過程也被稱為 兩步PCR法。當使用低合成能力的 Taq 聚合酶時,如Taq聚合酶,快速循環條件可能適用于 <500 bp左右的短片段。擴增如此大小的片段通常不需要延長聚合時間,因此可縮短PCR方案中的延伸步驟時間。為確定最短延伸時間,同時又不損失產物得率,可采用一系列延伸時間遞減的方式(幾秒)優化PCR。每個目的片段和引物對都可能會產生變化結果,所以需要在特定條件下對快速PCR進行優化。快速PCR的另一種調整方式是縮短變性時間,將變性溫度提高至98°C。使用這種策略時應注意,非高度熱穩定的酶在這種高溫環境下易于變性。表 .使用低合成能力DNA聚合酶實現快速PCR時所用的反應參數。
使用可實現快速循環的熱循環儀和薄壁 PCR管,分別有助于實現快速變溫和高效熱轉移,從而能夠大大加速PCR。直接PCR是指直接從樣品擴增目標DNA,無需進行核酸分離純化。直接PCR中,在高溫變性階段,諸如細胞、組織等材料在特殊的緩沖液中被裂解,釋放出DNA。因此這種方法簡化了實驗流程,減少了動手操作時間,同時可避免純化步驟DNA的損失。
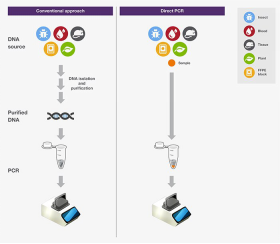
推薦使用具有高合成能力的DNA聚合酶用于直接PCR擴增。細胞碎片、蛋白、脂質和多糖也隨DNA一起被釋放到裂解液中,它們會抑制PCR反應。而具有高合成能力的DNA聚合酶能夠耐受這類抑制劑,使直接PCR擴增成為可能。具有高合成能力的酶通常具有更高的靈敏度,因此可從未純化的樣品中成功擴增微量DNA。具有高GC含量(>65%)的DNA模板由于G和C堿基間的強氫鍵影響,比較難以擴增。富含GC的序列同時也涉及二級結構。因此,富含GC的序列可導至DNA聚合酶沿模板擴增時“卡頓”并干擾DNA合成。為了擴增高GC含量的片段,雙鏈模板必須解離,以便引物與模板結合,并使DNA聚合酶能夠讀取到序列。為了克服強GC相互作用,最常用的方法是使用DMSO等PCR添加劑或輔助溶劑來幫助DNA變性。然而,這些試劑通常會降低引物的 Tm,所以退火溫度也需進行相應的調整。高合成能力的DNA聚合酶由于與模板的結合能力更強,有利于完成高GC含量PCR。超高熱穩定性DNA聚合酶也有利于高GC含量PCR,因為較高的變性溫度(如,使用98°C代替95°C)可能會促進雙鏈解離和PCR擴增。多重PCR可在同一PCR反應管中同時擴增多個不同的片段。多種PCR不僅意味著節省時間、試劑和樣品,還能夠同時對比多個擴增子。 當一個PCR管中有多個引物對時,如在多重PCR中,因無法僅針對一個引物對或目的片段進行反應優化,而是要考慮到所有引物和靶標,所以可能會出現非特異性擴增和效率降低。因此,為盡量減少由非特異性擴增導至的錯配,應對引物進行精心設計。首先,引物序列應盡可能與其目的序列一一對應,并且所有引物的 Tm相差不應超過5°C。在多重PCR開始前,應利用單個PCR反應驗證每個引物對的特異性和擴增效率。此外,擴增子應具有不同的大小,從而能夠通過凝膠電泳對其進行分離鑒定。除了引物設計和擴增子大小,使用熱啟動DNA聚合酶和專為多重PCR設計的緩沖液也將有助于獲得成功的PCR結果和提高反應特異性。 盡管多重PCR常作為終點法PCR,但由于其在多重標記和檢測中的能力 ,將其用于實時熒光定量PCR也變得越來越流行。另外,多重實時熒光定量PCR也常被用于遺傳標志物的檢測,用于人類身份鑒定。長片段PCR通常是指擴增大于5kb的DNA片段。長片段PCR傳統上使用 Taq DNA 聚合酶(用于快速延伸)和高保真酶(用于提高準確性)的混合物。隨著具有高合成能力的高保真DNA聚合酶被發明出來,現在能夠在更短的時間內實現更準確的長片段PCR。通過在DNA聚合酶中設計一個較強的DNA結合結構域,從而使其能夠在短時間內擴增長片段(如,來自gDNA的 >20
kb 片段),實現高合成能力。此外,極高的保真度(如, Taq 聚合酶保真度的 >100倍)還有助于確保長片段擴增的低錯誤率。
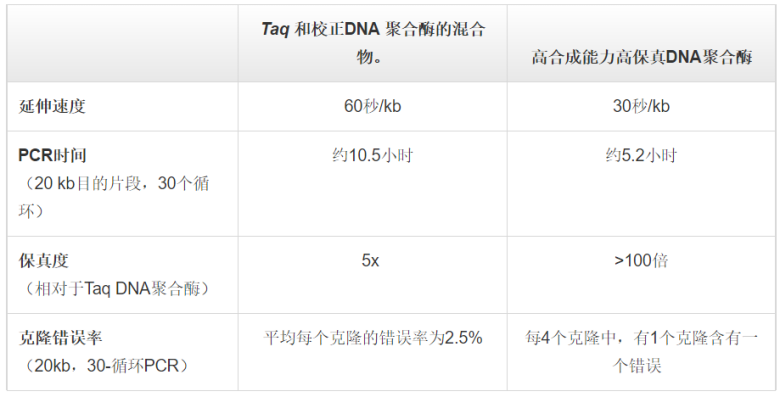
表 .在長片段PCR和克隆中使用高合成能力高保真DNA聚合酶的優勢。DNA聚合酶的高合成能力可顯著縮短長片段PCR的反應時間(在本例中,時間縮短了一半),而高保真度可減少篩選含正確插入片段克隆的工作量。當擴增>10 kb的目的片段時,應根據以下5個關鍵點對PCR方案進行優化:2. 如果DNA聚合酶的熱穩定性較低,則需要使用更多量的酶,以彌補因延長循環時間導至的活性損失。4. 適當延長PCR步驟的持續時間,有助于模板DNA的完全解離及引物的結合。5. 適當延長PCR延伸時間,可確保目標區域的全長復制。 反向PCR起初設計用于確定鄰近未知區域的序列。它有助于研究基因的啟動子序列;致癌性染色體重排,如基因融合、易位和轉座;以及病毒基因整合。該方法之所以被稱為反向PCR,是因為引物設計用于向兩邊延伸而不像常規PCR中朝著彼此延伸。如今,反向PCR常被用于定點突變,復制一個具有預期突變的質粒。在研究基因組DNA未知序列的傳統工作流程中,首先進行限制性酶切消化和連接,再進行反向PCR,隨后對PCR擴增子進行測序。對于gDNA消化,需選用一種限制性內切酶進行酶切,以獲得長度合適且能夠自我連接的片段。同時,選定的限制性內切酶不可剪切已知序列,從而使連接發生于側翼未知序列之間。使用低濃度的酶切DNA片段優化連接步驟,使其傾向于自我連接而非多片段連接(即形成連環體)。完成自我連接后,從DNA的已知區域啟動反向PCR。所獲得的擴增子每個末端都含有部分已知DNA序列。隨后,可從末端開始對這些擴增子進行測序,檢測上述已知序列的相鄰區域。序列的擴增程度(得率)取決于模板起始量,PCR常用于對樣品中的DNA進行定量,其中,最常見的應用是基因表達定量。終點PCR方法雖然可行,但它存在一重大缺點,即需要通過凝膠電泳確定得率,從而限制了檢測靈敏度。此外,定量是在PCR末期進行的,而此時的擴增已達到平臺期,因此,DNA凝膠染色強度無法與DNA起始量呈線性相關。盡管如此,若在到達平臺期之前通過終點PCR對基因表達進行半定量分析,可使用連續稀釋的DNA樣品作為起始物,或收集指定PCR循環的擴增子,并根據凝膠染色強度估計基因表達量。 直到1993年,Higuchi等報道稱使用熒光信號對PCR擴增進行實時監測,才克服了終點PCR定量的局限性。這一技術為我們今天所熟知的定量PCR(qPCR)奠定了基礎。1997年,第一款qPCR儀進入市場,使PCR能夠準確定量基因表達和拷貝數。qPCR依靠對指數期目的片段擴增熒光信號的實時監測,克服了終點PCR定量的缺點。盡管qPCR能夠定量檢測相對和絕對基因表達,但是其檢測能力限制了定量性能。20世紀90年代與實時熒光定量PCR同時開發的數字PCR (也稱為極限稀釋PCR)實現了真正的DNA樣品絕對定量。在數字PCR中,是將高度稀釋的DNA樣品分配到多區室芯片中,使每個區室最多含有一個拷貝的靶標。然后,對每個區室內的擴增進行檢測,獲得陽性或陰性結果(分別為1或0個模板拷貝;即, "數字"
結果)。最后,使用統計模型(泊松分布),根據陰性反應部分確定樣品的拷貝數,無需定量已知樣品(標準品)。除了基因表達和拷貝數定量,數字PCR還適用于區分低頻等位基因、病毒滴定以及下一代測序文庫的絕對定量等應用。利用數字PCR進行絕對定量的一般工作流程。 總之,改進的PCR實驗方案和改進的DNA聚合酶旨在改善PCR擴增的結果。雖然PCR的基礎概念并未發生改變,但新型PCR方法將繼續推動和簡化分子生物學研究。